作者:找药宝典
2022年和2023两年间,新药批准、新适应证和药物移除继续为血液系统恶性肿瘤患者增加了新的治疗选择,尽管血液系统恶性肿瘤的扩大适应证和新批准非常令人兴奋,但血液系统肿瘤仍有许多未满足的需求。目前临床上仍对新疗法有巨大需求,我们期待2023年看到更多令人期待的新疗法。本文中我们将回顾针对血液系统恶性肿瘤重点药物的最新批准,帮助大家更好的了解与掌握新兴疗法在血液系统肿瘤中的临床效果与使用准则。
01白血病
1.1 天冬酰胺酶途径
天冬酰胺对细胞存活过程非常关键,而天冬酰胺酶是急性淋巴细胞白血病(ALL)和淋巴母细胞淋巴瘤(LBL)多药治疗方案的核心组成部分天冬酰胺酶将血清天冬酰胺酶转化为天冬氨酸和氨,这反过来又阻止白血病细胞获得天冬酰胺。没有天冬酰胺,白血病细胞就不能合成RNA或蛋白质,也就不能增殖和存活。因此,维持门冬酰胺酶活性的时间是ALL和LBL治疗的关键。自1978年首次引入门冬酰胺酶以来,ALL的生存率估计值一直在稳步提高。
天冬酰胺酶(Rylaze,Recombinant)被指作为多药化疗方案的一部分,用于对大肠杆菌衍生的门冬酰胺酶有超敏反应的1个月或1个月以上的ALL/LBL成人和儿童患者。该药物于2021年6月在美国获批,给药方案为每48小时25 mg/m2。然而,这一给药方式对患者具有挑战性,尤其是因为输注中心在周末不开放。
许多接受这种治疗的患者是儿科患者,每隔一天来一次可能对他们和工作的父母来说都是不现实的。因此,2022年11月,FDA标签扩大,包括周一/周三/周五给药时间表。该方案是根据2/3期临床试验JZP458/ AALL1931的数据批准的,新的给药方案为周一上午和周三上午均为25 mg/ mg2,周五下午为50 mg/m2。在试验中,给药方案显示了良好的风险-获益特征,并且通过模拟,超过90%的患者达到了血清门冬酰胺酶最低活性大于0.1 U/mL。
1.2 IDH突变
约20%的急性髓系白血病(AML)患者可检测到IDH1或IDH2突变。因此,enasidenib (IDHIFA)于2017年被批准用于治疗idh2突变的复发/难治性(R/R) AML, ivosidenib (Tibsovo)于2018年被批准用于治疗idh1突变的R/R AML。2022年5月,Ivosidenib与阿扎胞苷联用也被批准用于有IDH1突变且不适合接受强化化疗的新诊断乳腺癌患者。本批准同时适用于不能耐受强化治疗的75岁及以上患者。
IDH1突变通常存在于7% ~ 14%的AML患者中。此外,骨髓增生异常综合征患者中约有3% ~ 4%存在这种情况。IDH1通过催化异柠檬酸氧化脱羧生成a-酮戊二酸发挥作用。
1.2.1 Olutasidenib
美国FDA于2022年12月批准奥鲁他尼布用于治疗复发/难治性AML。剂量为每日2次,每次150毫克。奥鲁他尼布可以穿过血脑屏障,这使得它不同于其他IDH1抑制剂。它已被纳入美国国家综合癌症网络(National Comprehensive Cancer Network, NCCN)指南,并且有一项正在进行中的研究将奥鲁他尼布与阿扎胞苷进行比较。
Olutasidenib是一种强效的、选择性的口服突变型IDH1小分子。这种基于喹啉酮的突变型IDH1变构非竞争性抑制剂结合在IDH1同型二聚体界面附近的疏水口袋中。它具有恢复正常细胞分化的治疗潜力。
Olutasidenib在一项2期试验中进行了评估,该试验纳入了携带IDH1突变的复发性或难治性AML患者。研究者为这153例idh1抑制剂treatment-naïve患者开出了每日2次、每次150 mg的奥鲁他尼单药治疗。本试验的主要终点是完全缓解加上外周血计数部分恢复的完全缓解(CR + CRh)率,结果为35% (n = 51;95% ci, 27.0% ~ 43.0%)。总有效率(ORR)为48% (n = 71;95% ci, 40.0% ~ 56.7%)。至CR/CRh的中位时间为1.9个月(0.9 ~ 5.6),中位缓解持续时间(DOR)为11.7个月(95% CI, 6.9 ~ 25.9),中位总生存期(OS)为11.6个月(95% CI, 8.9 ~ 15.5) 。
本试验中任何级别的最常见不良事件(ae)均为胃肠道相关事件,包括恶心(38%)、便秘(26%)和腹泻(20%)。
02淋巴瘤
以前有3种PI3K抑制剂,但现在只有1种仍在市场上。静脉给药的copanlisib (Aliqopa)仍获得批准,但idelalisib (Zydelig)和duvelisib (Copiktra)在长期确证性研究证明其获益/风险特征不利后,被从指南中删除,并失去了批准。
一种EZH2抑制剂他泽司他(tazemetostat, Tazverik)被批准用于R/R滤泡性淋巴瘤,以及两种cd19导向的CAR t细胞疗法。它们是axicabtagene ciloleucel (Yescarta)和tisagenlecleucel (Kymriah)。
2.1 Mosunetuzumab
于2022年底获批的Mosunetuzumab是首个在这种情况下获批的CD20/CD3 t细胞结合双特异性抗体,代表了一类新的固定疗程癌症免疫疗法。患者接受8个周期治疗。如果达到CR,则可以“停止观察”,但如果未达到CR,则可以在停止观察前继续治疗17个周期。它是一种现成产品,适用于已经接受过2线或2线以上全身性治疗的R/R滤泡性淋巴瘤成人患者。
双特异性抗体是如何工作?就好比它有一只手臂伸出去抓住目标细胞,然后它有一只手臂伸出去抓住你的CD3 T细胞,激活这些T细胞,然后找到并瞄准淋巴瘤细胞。
Mosunetuzumab获得批准的依据是一项2期、单组、多中心研究的结果,该研究共纳入了90例1 ~ 3a级滤泡性淋巴瘤患者,这些患者既往接受过≥2线治疗(包括CD20单克隆抗体和蒽环类药物)。中位随访18.3个月后,ORR为80% (95% CI, 70.3 ~ 87.7), CR率为60% (95% CI, 49.1% ~ 70.2%)。中位CR时间为3个月(1.4 ~ 5.7),中位无进展生存期(PFS)为17.9个月(95% CI, 10.1-未达到[NR])。中位缓解持续时间为22.8个月(95% CI, 9.7-NR)。
细胞因子释放综合征(CRS)是该疗法最令人担忧的问题。本试验的90例患者中有40例(44%)发生了CRS, 26%为1级严重程度,17%为2级严重程度。CRS多发生在第1周期的第1天(23%),也可发生在第15天。

2.2 Brentuximab vedotin
Brentuximab vedotin于2022年11月首次获得儿科批准。它被批准与多柔比星、长春新碱、依托泊苷、泼尼松和环磷酰胺联用,用于治疗≥2岁的未经治疗的高危经典霍奇金淋巴瘤患儿。
这项批准得到了一项随机、开放标签、积极对照3期试验结果的支持,该试验纳入了600例患者,分别接受维布妥昔单抗(brentuximab vedotin)或博来霉素(与多柔比星、长春新碱、依托泊苷、泼尼松和环磷酰胺联合治疗)。高危疾病的定义为Arbor分期ⅱb期伴大面积病变、ⅲb期、ⅳa期和ⅳb期。
在42.1个月(范围,0.1 ~ 80.9)随访时,brentuximab组和标准治疗组的3年无事件生存率(EFS)分别为92.1% (95% CI, 88.4% ~ 94.7%)和82.5% (95% CI, 77.4% ~ 86.5%) (HR, 0.41, 95% CI, 0.25 ~ 0.67)。两组均未达到中位EFS,但维布妥昔单抗组的3年OS率为99.3%,而标准治疗组为98.5%。据报道,两组的毒性特征相似。
2.3 Zanubrutinib
SEQUOIA研究是一项3期试验,在年龄较大、有合并症且疾病不携带高危基因组异常del(17)的患者中,评估了在未经治疗的CLL或SLL中,泽布替尼与利妥昔单抗和苯达莫司汀(R-benda)的比较。本试验是基于之前证明zanubrutinib在这一情况下具有优效性的研究。共纳入590例患者,其中241例接受赞布替尼治疗,238例接受R-benda治疗。中位随访时间为26.2个月.
总体而言,与R-benda相比,zanubrutinib显著改善了中位PFS(风险比,0.42;95%CI:0.28 - -0.63)。值得注意的是,两组均未达到独立审查委员会规定的中位PFS。zanubrutinib组的中性粒细胞减少发生率远低于R-Benda组(分别为11% vs 51%)。这些数据促使赞布替尼被批准用于CLL或SLL患者的一线治疗。
zanubrutinib有许多实际意义。对于化疗免疫治疗有困难的患者,它是一种口服治疗选择。此外,在ASPEN和ALPINE试验中,与伊布替尼治疗复发性心房颤动相比,泽布替尼与心房颤动低发生率(3%)相关。与在这种情况下批准的其他一些方案相比,它被认为毒性较小。
03多发性骨髓瘤
多发性骨髓瘤是一种不可治愈的恶性肿瘤,其特征是浆细胞失控增殖。这些恶性浆细胞会分泌m蛋白进入血液或尿液,与终末器官功能障碍有关。这是一种罕见的恶性肿瘤,但它是第二常见的血液系统恶性肿瘤。
然而,在多发性骨髓瘤的治疗方面看到了显著的增长,特别是自从硼替佐米(Velcade)在21世纪初获得批准以来。在获得批准之前,由于自体或异基因干细胞移植是唯一的标准治疗,因此估计5年相对生存率接近25.5%。然而,自那时起,许多药物有适应证,包括2015年以来的许多适应证。目前MM的5年相对生存率可以达到57.9%。
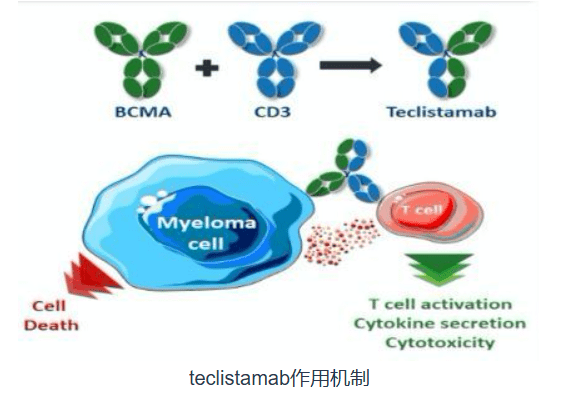
3.1 Teclistamab
MajesTEC-1试验是一项1/2期研究设计,纳入了165例既往未接受过bcma指导的治疗的R/R多发性骨髓瘤患者,其中77.8%患三级难治性疾病。患者接受每周一次1.5 mg/kg的teclistamab皮下给药,递进剂量分别为0.06 mg/kg和0.3 mg/kg。主要终点是ORR。次要终点包括PFS、OS、微小残留病(MRD)阴性和dor。
结果显示ORR为63%,包括58.8%非常好的部分缓解率和39.4%的CR率。中位DOR为18.4个月,至首次缓解的中位时间为1.2个月,至最佳缓解的中位时间为3.8个月。MRD阴性为26.7%,中位PFS为11.3个月,中位OS为11.3个月。根据这些数据,FDA批准将既往接受过至少4线治疗(包括抗cd38单克隆抗体、蛋白酶体抑制剂和免疫调节剂)的R/R多发性骨髓瘤患者用于治疗 。
NCCN指南针对这一情况添加了该指标。确证性3期试验仍在进行中。
关于teclistamab的安全性,最大的问题将是CRS。安全级别CRS发生率为72.1% (n = 119)。中位发病时间为2 d(1 ~ 6),中位持续时间为2 d(1 ~ 9)。
此外,由于CRS的风险和神经毒性,该药物仅在风险评估和缓解策略(REMS)下可获得,称为Tecvayli REMS。
3.2 Belantamab mafodotin
Belantamab mafodotin于2022年11月22日从美国市场下架。3期dream -3试验表明,该药物未达到主要终点pfs。
然而,belantamab mafodotin仍可通过同情用药获得,因此RNs和APPs应意识到该药物与角膜病变相关。接受该药治疗的患者应在基线时、每次给药前以及给药后出现黑麦眼、视力模糊等症状时立即接受眼科检查。还应建议患者使用不含防腐剂的润滑剂滴眼液,并避免佩戴隐形眼镜,除非在眼科医师的指导下。她强调,预防性使用类固醇滴眼液并不能预防或降低角膜病变的风险。如果发生角膜病变,应坚持治疗直至好转。根据严重程度,治疗可以恢复,也可以永久停止。
声明:本资料中涉及的信息仅供参考,请遵从医生或其他医疗卫生专业人士的意见或指导。
本文版权归找药宝典所有,任何个人或机构转载需获得找药宝典授权,在授权范围内使用,并标注来源“找药宝典”。
本文转载自其他网站,不代表健康界观点和立场。如有内容和图片的著作权异议,请及时联系我们(邮箱:guikequan@hmkx.cn)
本文来自投稿,不代表长河网立场,转载请注明出处: http://www.changhe99.com/a/vV6v4GXpwG.html
